Врожденные и наследственные заболевания новорожденных. Что такое врожденные болезни и можно ли с ними жить
Врожденные заболевания проявляются сразу после рождения, а наследственные могут выявить себя как сразу после рождения, так и спустя некоторое время - дни, месяцы и даже многие годы.
под наследственными заболеваниями понимают заболевания, которые передаются от родителей к детям. Причина этих болезней находится в генах (участках хромосом) или хромосомах половых клеток родителей. Эти мутации могут возникать вследствие ряда причин - радиационного облучения, употребления алкоголя и наркотиков, при инфекционных и вирусных заболеваниях и т. д. Особенно опасными для здоровья будущего ребенка являются мутации в яйцеклетках женщин
Доминантные и рецессивные наследственные заболевания относятся к моногенным, т. е. за их развитие отвечает изменение лишь одного определенного гена.
Примерами наследственных болезней, передающихся по доминантному типу, могут служить: арахнодактилия (см.), некоторые формы миопатии (см.), неврофиброматоз (см.), хондродистрофия (см.), наследственная атрофия слуховых нервов, туберозный склероз и др.
Если наследственная болезнь передается по аутосомно-рецессивному типу , то больные дети могут рождаться от внешне здоровых родителей, но являющихся носителями мутантного гена, так называемых гетерозиготных носителей. Аутосомно-рецессивный тип имеет особое значение в наследственной патологии, поскольку по этому типу передается большинство наследственных болезней обмена веществ: фенилкетонурия (фенилпировиноградная олигофрения), амавротическая семейная идиотия, галактоземия (см.), гепато-лентикулярная дегенерация (см.) и ряд других болезней.
В некоторых случаях проявляются наследственные болезни , вызываемые хромосомными мутациями (хромосомные болезни). расхождения хромосом в мейозе (см.), ведущее к появлению гамет, а затем и зигот с лишней хромосомой или, наоборот, с нехваткой ее, серьезно нарушает развитие. Организм, развившийся из такой зиготы, обычно гибнет на стадии зародыша или плода, но в ряде случаев выживает и становится носителем одной из тяжелых конституциональных аномалий, например болезни Дауна, синдрома Клайнфелтера или Шерешевского - Тернера, трисомии-Х и др. Проявляться наследственные болезни могут в любом возрасте. Многие наследственные болезни развиваются в детском возрасте, некоторые из них обнаруживаются при рождении или в первые дни жизни ребенка: гемолитическая болезнь новорожденных (см.), врожденная миотония (см.), галактоземия. На первом году жизни проявляется фенилкетонурия, спинальная амиотрофия, наследственные рахитоподобные синдромы, болезнь Ниманна - Пика и т. д. В раннем детском возрасте возникают и приобретают очерченную клиническую картину некоторые формы миопатий, туберозный склероз, гаргоилизм. Отдельные наследственные болезни проявляются в более позднем возрасте, иногда в старческом. Примером может служить хорея (см.) Гентингтона.
Диагностика производится цитогенетическими, молекулярно-цитогенетическими и молекулярно-генетическими методами, а также на основании фенотипическихпризнаков (синдромологический подход).
Однако имеется целый ряд специфических методов , с помощью которых можно изучить вопросы возникновения, развития, распространения, механизмы передачи из поколения в поколение наследственных болезней и роль генотипа и факторов среды в их проявлении.
Клиникогенеалогический метод . Он основан на построении родословных и прослеживании в ряду поколений передачи определенного признака. Этот метод относится к наиболее универсальным методам медицинской генетики.
Близнецовый метод изучения генетики человека позволяет определить роль генотипа и среды в проявлении признаков.
Цитогенетические методы . Основаны на макроскопическом исследовании кариотипа.
Биохимические методы . Основаны на изучении активности ферментных систем либо по активности самого фермента, либо по количеству конечных продуктов реакции, катализируемой данным ферментом. С помощью биохимических нагрузочных тестов можно выявлять гетерозиготных носителей патологических генов, например фенилкетонурии.
ВРОЖДЕННЫЕ болезни, нарушения структуры, функций и биохимии организма, обусловленные родовыми или дородовыми причинами и приводящие к физическим либо психическим отклонениям, болезни или смерти. К дородовым причинам таких пороков относятся наследственные факторы и(или) воздействия окружающей среды на развитие зародыша. Причиной возникновения пороков во время родов могут быть травмы или инфекции. Очень низкий вес при рождении, который отражает либо недоношенность, либо недостаточность процессов развития плода и является основной причиной детской смертности и инвалидности, тоже рассматривается как врожденный порок.
Хотя подавляющее большинство детей рождаются нормальными, примерно 1% имеют некоторые формы врожденных отклонений. На развитие ребенка в матке может повлиять огромное число факторов, однако многие отклонения можно исправить до или после рождения.
ВРОЖДЕННЫЕ ЗАБОЛЕВАНИЯ СЕРДЦА
Сердце - это сложный орган, значительная часть которого формируется в 3-7-ю неделю после зачатия. Врожденные заболевания сердца достаточно распространены и обнаруживаются у одного из сотни детей. Диапазон отклонений очень широк.
Мать с врожденным пороком сердца или уже родившая ребенка с таким заболеванием немного рискует в отношении следующего малыша. Многие заболевания сердца связаны с генетическими нарушениями, например синдромом Дауна; и если обнаружено заболевание сердца, может быть предложено обследование для выявления подобных проблем.
Как диагностируются заболевания сердца Большинство заболеваний выявляют с помощью ультразвукового обследования на 18-22-й неделе, поскольку многие дефекты ранее определить невозможно. Если в роду встречались заболевания сердца, то во время беременности должно проводиться регулярное ультразвуковое обследование. Однако некоторые заболевания ультразвук абсолютно не «ловит», и в целом примерно 40% болезней пропускается.
Лечение
В настоящее время развиваются методы хирургического внутриматочного лечения, потому в будущем некоторые отклонения станут устранять еще до рождения. Отношение к заболеванию после рождения будет зависеть от серьезности диагноза. Если проблема незначительная, то ребенок обычно остается с матерью под наблюдением педиатра больницы. Более серьезные заболевания, приводящие к недостатку кислорода в организме, требуют контроля специалиста. Это означает, что роды должны произойти тогда, когда посоветует доктор. Многие заболевания сердца подлежат хирургическому лечению, хотя имеются некоторые, которые не дадут ребенку возможность выжить вне матки.
Многоводие (гидрамнион) Это заболевание сердца у ребенка, при ультразвуковом обследовании кожа кажется опухшей, обнаруживается жидкость в грудной клетке и брюшной полости. Причины могут быть различные, в том числе несовместимость по группе крови, которая определяется при беременности.
Как диагностируется многоводие
Лечение
Лечение зависит от сложности состояния. В некоторых случаях, как анемия, лечение возможно, но в других, связанных с серьезными заболеваниями сердца, нет. Дети с несовместимой группой крови могут быть вылечены с помощью внутриматочного переливания крови. Выживет ли ребенок с многоводием, зависит от диагноза и
того, насколько он болен на момент установления диагноза. Очень больные дети не выживают.
Незаращение перегородки сердца
Обычно известно как «отверстие в сердце». Возникает при разделении ткани между меньшей и большей полостями сердца. Небольшое незаращение могут обнаруживать не сразу, а в дальнейшей жизни. Незаращения, определяемые во время беременности, обычно имеют большую величину или связаны с другими заболеваниями.
Как диагностируется незаращение
перегородки сердца
С помощью ультразвукового обследования.
Лечение
Небольшие дефекты не всегда требуют хирургического вмешательства, а в остальных случаях это необходимость.
Заболевания, связанные с течением крови от сердца
Возникают из-за того, что кровяные сосуды сообщаются неправильно или из-за отсутствия должного формирования клапанов. Заболевания подобного рода обычно являются сложными и могут быть связаны с незаращением перегородки сердца.
Как диагностируются заболевания, связанные с течением крови от сердца С помощью ультразвукового обследования.
Лечение
Если сердечные клапаны поражены, хирургическая операция является более сложной, и в долгосрочном плане вероятность успеха меньше. Многие подобные состояния представляют собой исключительную трудность. При наличии подобных отклонений у ребенка родителям следует проконсультироваться со специалистом во время беременности, чтобы выбрать наилучший вариант лечения.
Дефекты нервной трубки
Одно из наиболее частых заболеваний, дефект нервной трубки представляет собой неспособность головного и спинного мозга развиваться
нужным образом в течение первых четырех недель беременности. Заболевание встречается у одного из 2500 детей, проживающих в Великобритании, и приводит к различной степени врожденных пороков. В целом оно регистрируется в значительно большем числе беременностей, но родители предпочитают прервать ее, если болезнь определяется на раннем этапе.
Расщелина позвоночника без спинномозговой грыжи является самой легкой формой заболевания, при котором нет поражения спинного мозга или его оболочек и один-два позвонка сформированы неправильно. При этой форме спинной мозг покрыт кожей, поэтому проблем обычно не возникает. Иногда болезнь обнаруживается только при рентгеновском обследовании в процессе жизни. Иногда вокруг места проявления болезни вырастает пучок волос или кожа имеет ямочки.
Миеломенингоцеле является более тяжелой формой расщелины позвоночника. В этом случае позвоночник имеет повреждение, иногда достигающее размера апельсина, куда попадают нервные ткани, мышцы и спинномозговая жидкость. Дефект приводит к повреждению нервов, в результате чего возникают проблемы с управлением мышцами, мочевым пузырем и кишечником. С этим связана гидроцефалия.
Анэнцефалия (врожденное отсутствие головного мозга) - самый серьезный порок нервной трубки. Отверстие в верхней части трубки приводит к частичному отсутствию формирования черепа и головного мозга. После рождения такие дети не выживают.
Как диагностируются дефекты нервной трубки
Расщелина позвоночника может обнаруживаться на 16-й неделе с помощью исследования серозной жидкости очень эффективно для выявления детей со значительными дефектами нервной трубки.
Лечение
Зависит от типа и размера дефекта нервной трубки и его серьезности, что определяется с помощью ультразвукового обследования и томографии после родов. Если у ребенка явный дефект, то это потребует операции на спинном мозге. Хотя операция устранит порок, она не восстановит нервы, которые не смогут развиться должным образом. Гидроцефалия также требует операции для облегчения состояния ребенка после рождения.
Меры предупреждения
Хотя причина дефектов нервной трубки неясна, в настоящее время есть очевидные доказательства того, что фолиевая кислота, витамин, обнаруживаемый в листовых овощах, должна присутствовать в организме на начальном этапе беременности, чтобы дать возможность позвоночнику закрыться должным образом. Так как только питанием трудно добиться поступления рекомендуемых доз, советуют в течение трех месяцев до зачатия и до 12-й недели беременности принимать препарат фолиевой кислоты. Специалисты предполагают, что все женщины детородного возраста должны потреблять ежедневно 400 микрограммов фолиевой кислоты. Матери, у которых предыдущий ребенок был с расщелиной позвоночника или анэнцефалией или принимающие некоторые лекарства, например для лечения эпилепсии, должны получать повышенные дозы фолиевой кислоты - 5 мг. Ее можно выписать у врача или приобрести в аптеках и супермаркетах.
Гидроцефалия
Это накопление спинномозговой жидкости в голове, вызываемое закупоркой дренажной системы вокруг головного мозга. Иногда голова ребенка становится очень большой. Преждевременные роды служат наиболее частой причиной гидроцефалии из-за повышенного риска кровотечения в мозг, которое может предотвратить поглощение спинномозговой жидкости. Гидроцефалия может также возникать у детей с врожденными пороками, например с расщелиной позвоночника, в некоторых случаях заболевания наследуется, иногда оно возникает вследствие инфекций. Если известно, что у ребенка подобное состояние, может быть рекомендовано кесарево сечение. Насколько серьезно болен ребенок, зависит от причины заболевания. Одни дети вырастают с нормальным интеллектом, другие могут иметь серьезные отклонения, однако до рождения этого предсказать нельзя.
376 ПОСЛЕРОДОВОЙ УКАЗАТЕЛЬ
Как диагностируется гидроцефалия Во время беременности гидроцефалию определяют с помощью ультразвукового обследования. После рождения измерения головы, проводимые у каждого новорожденного, могут показать существующее состояние. Ранняя диагностика и лечение обеспечивают лучший результат.
Лечение
После рождения ребенка обычно проводится операция для отвода спинномозговой жидкости через шунт в кровоток. Шунт остается на всю жизнь. Операция по введению временного шунта может иногда выполняться до рождения. Постоянный шунт устанавливается после рождения. Для лечения некоторых форм гидроцефалии с помощью современных хирургических методов в черепе выполняется отверстие.
Церебральный (корковый) паралич
Термин относится к группе нарушений, касающихся движения и осанки. У одного из четырех детей наблюдаются связанные с заболеванием трудности с обучением. Причиной болезни может быть ненормальное развитие мозга до рождения, кислородная недостаточность, инфекция, кровотечение в мозге или родовая травма. Физические симптомы варьируются от слабости и вялости мышц до мышечной спастичности и неподвижности.
Как диагностируется церебральный паралич Обычно нельзя сделать точный диагноз, пока ребенку не исполнится год, так как многие части нервной системы до этого момента еще не полностью развились. Диагностика может осуществляться с помощью электроэнцефалографии, компьютерной томографии, а также проверок зрения и слуха. В некоторых случаях делают анализ крови для оценки наследственного влияния.
Лечение
Церебральный паралич не лечится, но есть виды лечения, которые помогают минимизировать последствия и развить способности ребенка. Среди них можно назвать физиотерапию, функциональную терапию и медикаментозное лечение. Для решения проблем деформированных конечностей иногда используется хирургическое вмешательство.
ЗАБОЛЕВАНИЯ МОЧЕПОЛОВОЙ СИСТЕМЫ Непроходимость мочевых путей
Непроходимость возникает, когда поток мочи между почками и мочевым пузырем блокируется частично или полностью, приводя к гидронефрозу (опуханию почек), что может стать причиной прекращения работы почек. Данное заболевание обнаруживается у плода уже на 15-й неделе. Гидронефроз слабой степени, также известный как почечно-тазовая дилатация, может самопроизвольно проходить к концу беременности и не требует лечения.
Как диагностируется непроходимость мочевых путей
Во время беременности заболевание определяется с помощью ультразвука. У новорожденных проводится ультразвуковое обследование почек с целью определения степени закупорки мочевых путей. Возможно проведение других обследований для выявления работоспособности почек.
Лечение
Хирургическое вмешательство по отношению к плоду выполняется только в очень тяжелых случаях, когда заболевание охватило обе почки. Операция эффективна, если сделана до того, как развивающиеся почки серьезно не повреждены. Сразу после рождения ребенка может потребоваться оперативное вмешательство, чтобы снять непроходимость. Для борьбы с инфекцией мочевых путей ребенку прописываются антибиотики.
Поликистоз почек
Когда почка плохо соединяется с выделительной системой, это может привести к ее плохому функционированию или отсутствию работоспособности, что на снимке ультразвукового обследования проявляется увеличением размера и наличием кист. Человек может жить с одной работоспособной почкой, а для развития в матке почки ребенку не нужны совсем, так как продукты распада удаляет плацента. Однако как только ребенок родится, ему необходимо, чтобы работала хотя бы одна почка. Иногда порок развития затрагивает только одну почку и у ребенка не будет впоследствии никаких серьезных проблем, при этом порой плохое функционирование исчезает в детском возрасте. Но если заболевание коснулось обеих почек, количество жидкости вокруг ребенка будет уменьшаться, и легкие не смогут нормально развиваться. После рождения ребенку станет трудно дышать, при этом и почки будут плохо функционировать. По этой причине двусторонний поликистоз почек приводит к летальному исходу.
Состояние, называемое «зрелый поликистоз почек» и наблюдаемое у взрослых, встречается и у плода. Он не вызывает никаких сложностей, пока организм не станет зрелым. Заболевание обычно наследуется от одного из родителей, хотя последний может и не знать об этом.
Как диагностируется поликистоз почек Во время беременности поликистоз определяют с помощью ультразвука. У новорожденных проводится ультразвуковое обследование почек, для проверки наличия кист применяется компьютерное сканирование.
Лечение
В зависимости от тяжести заболевания может потребоваться операция, когда ребенок станет старше. Если болезнь носит наследственный характер, могут потребоваться дополнительные анализы.
Гипоспадия
Заболевание обнаруживается примерно у одного из 300 мальчиков. Отверстие мочеиспускательного канала, представляющего собой трубку, через которую моча выводится из организма, и нормально располагающегося на конце полового члена, открывается в другом месте, обычно на нижней поверхности члена. Следовательно, возникают проблемы с мочеиспусканием, при этом член может быть изогнут вниз, что по достижении половой зрелости может повлиять на сексуальную жизнь.
Как диагностируется гипоспадия
♦ Невозможность нормального потока мочи.
♦ Изогнутый половой член.
♦ Закрытая крайняя плоть.
Лечение
В очень легких случаях не стоит предпринимать никаких действий. В более серьезных необходима хирургическая операция для продления мочеиспускательного канала. Дети с гипоспадией не должны подвергаться обрезанию, так как крайняя плоть при операции может использоваться в хирургических целях.
Неопущение яичка
При нормальном развитии плода яички по каналу опускаются из живота в мошонку. В некоторых случаях до рождения этого не происходит, почему, точно неизвестно. Данное заболевание относительно часто встречается у недоношенных детей, у выношенных - значительно реже. Яички обычно опускаются на 28-й неделе беременности, поэтому если ребенок рождается до этого срока, у яичек может не быть времени, чтобы спуститься. Иногда опускается только одно яичко, иногда опущение происходит не полностью.
На что следует обращать внимание
♦ Мошонка кажется маленькой или яички разной формы.
♦ Яички в мошонке не ощущаются.
Лечение
Обычно яички опускаются сами по себе в течение первого года. Иногда яичко, находящееся в паховом канале и опустившееся не до конца, может встать на место самопроизвольно. Если яичко не опускается, возможно лечение гормонами, чтобы помочь ему сделать это, или требуется хирургическая операция.
Если заболевание не лечить, у ребенка появляются высокие шансы остаться бесплодным и развитие рака яичка в зрелом возрасте.
ЗАБОЛЕВАНИЯ ПИЩЕВАРИТЕЛЬНОГО
ТРАКТА
Непроходимость кишечника
Непроходимость может возникать в любом месте кишечника, от пищевода до ануса. Закупорка в верхней части может привести к накапливанию амниотической жидкости, что обычно диагностируется во время беременности. Закупорка сразу после желудка называется дуоденальная атрезия. Этот тип непроходимости обычно обнаруживают у детей с синдромом Дауна, поэтому понадобится обследование. Закупорка в нижних отделах кишечника до рождения не определяется.
Как диагностируется непроходимость
кишечника
Для определения причины непроходимости
применяется ультразвуковое обследование.
Лечение
В случае непроходимости ребенку после рождения требуется хирургическая операция, чтобы обойти заблокированное место и дать возможность ребенку питаться.
Дефекты брюшной стенки
Дефекты возникают, когда часть брюшной стенки остается недоразвитой и в ней возникает отверстие. Содержимое брюшной полости выпадает наружу. В некоторых случаях имеется оболочка, в которой находится содержимое. Такое заболевание называется омфалоцеле (эмбриональная грыжа) или пупочная грыжа, оно может быть связано с другими генетическими проблемами ребенка; в этом случае предлагается обследование. Если кишечник не закрыт, состояние называется гастросхизис (врожденный мышечный дефект в стенке живота, обычно с выпячиванием внутренних органов, но не в области пупочного кольца). Дефект не связан с какими бы то ни было проблемами развития ребенка. Если ребенок остается здоровым, возможны вагинальные роды. Иногда, если у ребенка наблюдаются другие отклонения или пупочная грыжа слишком большая, предлагается кесарево сечение.
Как диагностируются дефекты брюшной стенки
Ребенок с подобным дефектом скорее всего родится маленьким, поэтому во время беременности требуется тщательный мониторинг. Дефекты брюшной стенки обычно выявляются при ультразвуковом обследовании.
Лечение
Для устранения дефекта ребенку требуется операция. Обычно достаточно одной. Иногда требуется удаление части кишечника, если она повреждена или закупорена. Кормление необходимо вводить постепенно, и ребенку потребуется достаточно продолжительное время (от двух до четырех недель), пока оно станет возможным.
Пилоростеноз
Заболевание возникает, когда привратник (кольцо мышц, связывающее двенадцатиперстную кишку с желудком) сужается до такой степени, что пища не может проходить. Симптомы обычно проявляются между 3-12-й неделями.
Болезнь чаще встречается у мальчиков и, как считают, имеет наследственный характер.
Как диагностируется пилоростеноз
Для подтверждения диагноза проводится физическое и ультразвуковое обследования.
Лечение
Заболевание устраняется простой хирургической операцией, при которой в привратнике выполняется небольшой разрез. После операции ребенок может нормально питаться и будет быстро набирать вес.
Диафрагмальная грыжа
Диафрагма представляет собой мышцу, отделяющую органы брюшной полости от органов грудной клетки. На раннем этапе развития плода в матке в диафрагме имеется отверстие, которое обычно закрывается к концу третьего месяца. Если отверстие остается открытым, содержимое брюшной полости, например кишки, может оказаться в грудной полости, и, заняв там место, препятствовать нормальному развитию легких и сердца.
Как диагностируется диафрагмальная грыжа
С помощью ультразвукового обследования. Дети с данным заболеванием после рождения часто имеют проблемы с дыханием.
Лечение
Для лечения диафрагмальной грыжи может потребоваться операция, чтобы закрыть дефект диафрагмы. Однако в тяжелых случаях маленький размер легких не дает ребенку возможности выжить. Надежные способы прогнозирования, определяющие до рождения достаточный размер легких, пока отсутствуют.
После успешной операции по устранению грыжи считается, что ребенок будет вести нормальную жизнь. Иногда диафрагмальная грыжа связана с генетическими заболеваниями, в этих случаях предлагается тестирование, чтобы устранить причины.
ЗАБОЛЕВАНИЯ КОСТНО-МЫШЕЧНОЙ
СИСТЕМЫ
Расщепленная губа или расщелина неба
Достаточно часто распространенные у младенцев дефекты, могут появляться как по отдельности, так и вместе. Порок возникает, когда верхняя губа плода и (или) небо не соединяются должным образом до рождения. Во многих случаях причина заболевания неизвестна, хотя иногда дефект может быть наследственным. Если дефект сложный, возникают трудности с кормлением.
Как диагностируется расщепленная губа или расщелина неба
В большинстве медицинских учреждений со специальным ультразвуковым оборудованием данные заболевания определяются до родов во втором триместре при обследовании. В противном случае дефект выявляется после рождения.
Лечение
Расщепленная губа устраняется хирургически у ребенка в возрасте трех месяцев. Расщепленное небо лечится в возрасте 6-15 месяцев. До этого момента в небо ребенка вставляется пластина, если возникают проблемы с кормлением. Пластическая хирургия дает хорошие результаты и обеспечивает нормальное развитие речи.
Деформированная стопа
У некоторых детей обнаруживается состояние, именуемое «косолапость». Заболевание встречается примерно у одного ребенка из тысячи. При этом одна или обе стопы отклоняются от нормального положения. Иногда болезнь вызывается генетическими причинами, но чаще она появляется из-за того, что стопа была зажата в матке, или потому, что кости ее не развились нормальным образом.
Как диагностируется деформированная стопа
Стандартное ультразвуковое обследование обычно отражает любые дефекты конечностей. При дальнейших проверках состояние уточняется.
Лечение
Если причиной является ограниченное пространство матки, то единственное, что потребуется после рождения, - это физиотерапевтические упражнения для выпрямления стопы. В случае недоразвития костей в детском возрасте ребенку придется сделать хирургическую операцию.
Заболевания позвоночника
Иногда часть позвонка неправильно сформирована или отсутствует, что дает искривление позвоночника. Порой это происходит вследствие генетических причин или хромосомных проблем. В таких случаях очень трудно предполагать, что будет в долгосрочном плане.
Как диагностируются заболевания позвоночника
С помощью ультразвукового обследования.
Лечение
Лечение зависит от сложности состояния. У некоторых детей проявляется ярко выраженный сколиоз (искривление позвоночника), который может быть устранен хирургическим путем.
Врожденный вывих бедра
Встречается относительно часто, связан с тем, что неправильно сформировавшийся бедренный сустав дает возможность головке бедра выскакивать из впадины тазовой кости. Причина вывиха бедра неизвестна, она может быть и наследственной. Заболевание у девочек встречается в десять раз чаще и чаще поражает детей при первых родах и при ягодичном предлежании. Часто встречается в случае двойни, а также может быть связано с такими врожденными состояниями, как расщелина позвоночника или синдром Дауна. Наиболее распространен вывих левого бедра, но в 25% наблюдается вывих обоих бедер.
Как диагностируется вывих бедра Во время обычного обследования новорожденного педиатр различным образом сгибает ножки ребенка. Щелканье может быть признаком врожденного вывиха бедра, тогда в 6-недельном возрасте проводится более тщательное обследование для проверки диагноза. К другим симптомам относятся асимметричные складки кожи на верхней части ножек и неспособность ребенка их вытянуть, расставив, например, при смене подгузника.
Лечение
Лечение на раннем этапе с помощью приспособления для фиксации повышает шансы нормального развития бедра. При смене подгузника приспособление легко снимается, оно не мешает кормлению, купанию и сну. Если лечение требуется после 6 месяцев, может использоваться гипс. Очень редко для увеличения впадины в тазовой кости требуется хирургическая операция. Обычно она осуществляется до того, как ребенок начнет ходить.
На начало 21 века насчитывается уже более 6 тыс. видов наследственных заболеваний. Сейчас во многих институтах мира изучаются человека, список которых огромен.
Мужское население имеет все больше генетических дефектов и все меньше шансов зачать здорового ребенка. Пока неясны все причины закономерности развития пороков, однако можно предположить, что в ближайшее 100-200 лет наука справится с решением этих вопросов.
Что такое генетические болезни? Классификация
Генетика как наука начала свой путь исследования с 1900 года. Генетическими болезнями называют те, которые связаны с отклонениями в генной структуре человека. Отклонения могут происходить как в 1 гене, так и в нескольких.
Наследственные болезни:
- Аутосомно-доминантные.
- Аутосомно-рецессивные.
- Сцепленные с полом.
- Хромосомные болезни.
Вероятность при аутосомно-доминантном отклонении - 50 %. При аутосомно-рецессивном - 25%. Болезни, сцепленные с полом, это те, что несет с собой поврежденная Х-хромосома.
Наследственнные заболевания
Приведем несколько примеров болезней, согласно вышеозвученной классификации. Итак, к доминантно-рецессивным заболеваниям относятся:
- Синдром Марфана.
- Пароксизмальная миоплегия.
- Талассемия.
- Отосклероз .
Рецессивные:
- Фенилкетонурия.
- Ихтиоз.
- Другие.
Сцепленные с полом заболевания:
- Гемофилия.
- Мышечная дистрофия.
- Болезнь Фарби.
Также на слуху хромосомные наследственные заболевания человека. Список хромосомных аномалий следующий:
- Синдром Шэрешевского-Тернера.
- Синдром Дауна.
К полигенным заболеваниям относятся:
- Вывих бедра (врожденный).
- Пороки сердца.
- Шизофрения.
- Расщепление губы и неба.
Самая распространенная генная аномалия - синдактилия. То есть срастание пальцев. Синдактилия - самое безобидное нарушение и лечится благодаря хирургии. Однако это отклонение сопровождает и другие более серьезные синдромы.
Какие заболевания наиболее опасны
Из тех перечисленных заболеваний можно выделить наиболее опасные наследственные заболевания человека. Список их складывается из тех видов аномалий, где в хромосомном наборе встречается трисомия или полисомия, то есть когда вместо пары хромосом наблюдается присутствие 3, 4, 5 или более. Встречается и 1 хромосома вместо 2. Все эти отклонения происходят из-за нарушения деления клеток.
Самые опасные наследственные заболевания человека:
- Синдром Эдвардса.
- Спинальная мышечная амиотрофия.
- Синдром Патау.
- Гемофилия.
- Другие заболевания.
Вследствие таких нарушений, ребенок проживает год или два. В некоторых случаях отклонения не столь серьезны, и ребенок может дожить до 7, 8 или даже до 14 лет.
Синдром Дауна
Синдром Дауна передается по наследству, если один или оба родителя являются носителями дефектных хромосом. Если точнее, синдром связан с хромосомы (т. е. 21 хромосомы 3, а не 2). Дети с синдромом Дауна имеют косоглазие, складку на шее, аномальную форму ушей, проблемы с сердцем и умственную отсталость. Но для жизни новорожденных опасности хромосомная аномалия не несет.
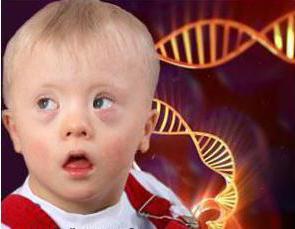
Сейчас статистика утверждает, что из 700-800 детей 1 рождается с этим синдромом. У женщин, которые хотят завести ребенка после 35, больше вероятность родить такого малыша. Вероятность где-то 1 к 375. А вот женщина, решившаяся на рождение малыша в 45, имеет вероятность 1 к 30.
Акрокраниодисфалангия
Тип наследования аномалии - аутосомно-доминантный. Причина синдрома - нарушение в 10 хромосоме. В науке эта болезнь называется акрокраниодисфалангия, если проще, то синдром Аперта. Характеризуется такими особенностями строения тела, как:
- брахикефалия (нарушения соотношения ширины и длины черепа);
- срастание коронарных швов черепа, вследствие этого наблюдается гипертензия (повышенное кровяное давление внутри черепа);
- синдактилия;
- выпуклый лоб;
- часто умственная отсталость на фоне того, что череп сдавливает мозг и не дает расти нервным клеткам.

В наше время детям, имеющим синдром Аперта, назначают хирургическую операцию по увеличению черепа, чтобы восстановить кровяное давление. А психическую недоразвитость лечат стимуляторами.
Если в семье есть ребенок, у которого диагностирован синдром, вероятность того, что 2 ребенок родится с тем же отклонением, очень велика.
Синдром счастливой куклы и болезнь Кэнэвэн — ван-Богарта — Бертрана
Рассмотрим подробнее и эти заболевания. Опознать синдром Энгельмана можно где-то с 3-7 лет. У детей бывают судороги, плохое пищеварение, проблемы с координацией движений. У большинства из них косоглазие и проблемы с мышцами лица, из-за чего на лице очень часто улыбка. Движения ребенка очень скованы. Для врачей это понятно при попытках ребенка ходить. Родители в большинстве случаев не знают, что происходит и тем более с чем это связано. Чуть позже заметно и то, что они не могут говорить, лишь стараются что-то нечленораздельно пробормотать.

Причина, по которой у ребенка проявляется синдром - это проблема в 15 хромосоме. Встречается заболевание крайне редко - 1 случай на 15 тыс. рождений.
Другая болезнь - болезнь Кэнэвэн - характеризуется тем, что ребенок имеет слабый мышечный тонус, у него проблемы с проглатыванием пищи. Заболевание обусловлено поражением центральной нервной системы. Причина - поражение одного гена в 17 хромосоме. Вследствие этого нервные клетки головного мозга разрушаются с прогрессирующей быстротой.

Признаки болезни можно увидеть в 3-хмесячном возрасте. Проявляется болезнь Кэнэвэн так:
- Макроцефалия.
- Судороги появляются в месячном возрасте.
- Ребенок не способен вертикально держать голову.
- После 3 месяцев повышаются сухожильные рефлексы.
- Многие дети к 2 годам слепнут.
Как видим, очень разнообразны наследственные заболевания человека. Список, приведенный лишь для примера, далеко не полный.
Хотелось бы отметить, что если у обоих родителей есть нарушение в 1 и том же гене, то шансы родить больного ребенка велики, но если аномалии в разных генах, то можно не опасаться. Известно, что в 60 % случаях хромосомные аномалии у зародыша приводят к выкидышу. Но все же 40 % таких детей рождаются и борются за свою жизнь.
Муниципальное общеобразовательное учреждение средняя общеобразовательная школа №1.
Проект
Тема: «Врожденные и наследственные детские заболевания».
Выполнила:
Ученица 11 класса
Гребенщикова Екатерина.
Проверила:
Учитель ОП
Кремнева Ирина Николаевна.
Г.о.Тольятти, 2009 гг
I . Введение ……………………………………………………………………………………….стр. 4
II . Основная часть:
1. Характеристика заболеваний…………………………………………………………….стр.5
2. Наследственные заболевания:
● ахондроплазия; ………………………………………………………………………………….стр.7
● гемолитическая болезнь новорожденных; …………………………………………стр.8
● микроцефалия; ………………………………………………………………………………….стр.8
● врожденные пороки сердца; ………………………………………………………………стр.9
3. Врожденные заболевания:
● альбинизм; ………………………………………………………………………………………..стр.12
● гемофилия; ………………………………………………………………………………………..стр.13
● синдром Дауна; ………………………………………………………………………………….стр.14
● олигофрения; …………………………………………………………………………………….стр.15
● детский церебральный паралич; ………………………………………………………..стр.18
● «заячья губа» и «волчья пасть»; ………………………………………………………..стр.22
III . Вывод ……………………………………………………………………………………………стр.25
Цель:
Ознакомиться с наследственными болезнями и болезнями передающимися потомству по наследству.
Проблемный вопрос:
Какие наследственные и врожденные болезни передаются потомству от родителей и обусловлены изменением наследственной информации?
Задачи:
1. Исследовать выбранную тему.
2. Выявить ряд заболеваний, при наличии которых противопоказана беременность для женщины.
Введение.
Здоровье вашего будущего ребенка зависит от вашего здоровья и здоровья отца ребенка. Будущим родителям желательно заранее, еще до наступления беременности, выяснить, можно ли им иметь детей, способны ли они произвести на свет здоровое потомство. Если это необходимо, то молодым супругам могут предложить пройти медицинское обследование.
В наше время здоровье молодых людей вызывает серьезную озабоченность, так как значительная часть из них имеют различные заболевания, и в частности такие, при которых возникает необходимость отказа от возможности рождения ребенка.
П
оявление на свет ребенка с врожденными дефектами развития всегда ошеломляет семью; эта тема - одна из самых тяжелых в акушерстве. Супруги в первый момент испытывают ни с чем не сравнимый психологический шок, который затем переходит в чувство вины, им кажется, что у них уже никогда не будет здорового ребенка.
Следует сразу сказать, что ребенок с врожденными пороками может появиться на свет абсолютно в любой семье - молодой, здоровой, без вредных привычек, с нормально протекающей беременностью. По данным многолетней статистики, во всем мире около 5% детей рождается с врожденными заболеваниями.
Врожденные пороки развития плода можно разделить на две большие группы - наследственно обусловленные
(то есть заложенные в генах и хромосомах, передающиеся по наследству) и собственно врожденные
(приобретенные в ходе внутриутробного развития). Такое деление довольно условно, так как большинство дефектов развития вызываются сочетанием наследственной предрасположенности и неблагоприятного внешнего воздействия, представляя собой мультифакториальные
аномалии.
Проблема врожденных пороков развития плода очень многообразна, изучением этого вопроса занимаются различные специалисты - генетики, неонатологи, эмбриологи, специалисты по дородовой (пренатальной) диагностике. Разобраться в причинах всегда бывает непросто.
С уществует такая поговорка: "Все мы стоим на плечах наших предков". Это утверждение верно не только в отношении культурного уровня, семейных традиций и воспитания. Малыши несут в себе генетическую информацию всех предыдущих поколений и, к сожалению, не всегда полезную. Что же нужно знать будущим родителям о наследственных болезнях? Наследственными называют те болезни, единственной причиной которых явилась "поломка", т.е. мутация гена. Наследственная предрасположенность может проявиться и с возрастом: для того чтобы дефектный ген "ожил", определенное сочетание внешних факторов должно его к этому подтолкнуть.
Характеристика заболеваний
Медицинские выгоды ношения ребенка
Врожденные болезни вызываются различными вредными факторами, которые действуют на будущую маму во время беременности. Хромосомные болезни происходят от нарушения структуры множества генов и хромосом. Мультифакторные заболевания (болезни, имеющие несколько причин) проявляются, если на малышей с определенным типом иммунной системы (прежде всего аномальным) действуют вирусы, бактерии, простейшие, токсические и лекарственные средства. Как правило, наследственные болезни проявляют себя лишь пороками развития разных органов и задержкой развития. Хромосомные дефекты - это куда более серьезная проблема, ведь они вызывают грубые интеллектуальные дефекты, такие, как, например, синдром Дауна. Многие наследственные болезни дебютируют в детском возрасте, хотя на практике могут быть обнаружены многие годы спустя. Они весьма разнообразны, точнее, у каждого заболевания есть своя генетическая основа. Сегодня расшифрованы боле 5 000 генов и свыше 60 000 последовательностей компонентов генов. Выделены наследственные "причины" таких бичей человечества, как сахарный диабет, атеросклероз, а также некоторых онкологических и инфекционных болезней. Перед международным научным сообществом стоит вполне реальная задача 0 в ближайшее время расшифровать все гены человека, ведь в состав 23 пар его хромосом входят "всего" 100 000 генов.
Как наследуются болезни?
Они передаются непосредственно от родителей к детям, но в этом случае плод погибает еще до рождения. Чаще наследование происходит иначе: болеют только дети, получившие дефектный ген от обоих родителей. Ели ребенку от родителей достаются разные гены - один здоровый, другой - больной, то болезнь никак не будет проявлять себя. Вероятность рождения больного ребенка у родителей, которые имеют и здоровый и больной ген, составляет 25%. Для болезней, "сцепленных с полом", т.е. контролируемых генами, которые находятся в половой хромосоме, характерно то, что ими болеют преимущественно мальчики, а носителями болезни являются девочки. Хромосомные заболевания, или синдромы, передаются по наследству, но не всегда, хотя генетически они предопределены.
Как распознать угрозу?
Любое заболевание важно вовремя обнаружить, в том числе и наследственное. Еще лучше - попытаться его избежать, т.е. обратиться за советом к врачу-специалисту. Наследственные болезни изучают врачи- генетики, и получить у них консультацию можно в медико-генетических центрах и центрах планирования семьи.
Чем могут помочь врачи?
В арсенале врачей есть различные методы определения и предотвращения наследственных и врожденных болезней. Пренатальная диагностика , которая позволяет узнать, каким будет наслество ребенка, еще до его рождения. Современный уровень дородовой диагностики немыслим без использования живых клеток и тканей, в том числе клеток крови плода, которые необходимы для изучения вещества наследственности - дезоксирибонуклеикновой кислоты (ДНК). Скринирующие (т.е. просеивающие) программы. В них участвуют семьи из групп риска. Дородовой патронаж . Очень важно, чтобы во время беременности будущая мама не подвергалась различным воздействиям, которые могли бы сказать на здоровье ребенка. Существует прямая и непрямая ДНК-диагностика наследственных болезней. Главное преимущество прямого метода - точность диагностики и отсутствие необходимости обследовать всю семью, т.к. объектом анализа является ген ребенка, а точнее мутации гена. Так выявляют тяжелые наследственные болезни, такие как миодистрофия Дюшенна, гемофилия А. Непрямые (косвенные) методы требуют обследований родителей и родственников больного ребенка с тем, чтобы выявить наследственные болезни, "гены-виновники" которых четко не определены.
Наследственные заболевания
Ахондроплазия.
Ахондроплазия является генетическим заболеванием, передающимся по наследству. Ахондроплазия приводит к тому, что у больного человека развиваются непропорционально короткие конечности. Эта генетическая аномалия чрезвычайно редка. Средняя высота взрослого мужчины, больного ахондроплазией, составляет всего 131см, средний рост у женщин с этим генетическим недугом 124 см.
Ахондроплазия является наиболее распространенной причиной аномалии в развитии человека, при котором конечности носителя такой аномалии вырастают непропорционально короткими.
Ахондроплазия в общем смысле является нарушением роста костей. Хотя ахондроплазия буквально означает "без формирования хрящей", проблема при заболевании ахондроплазией не в том, что хрящи у больного этой аномалией не формируются, а в преобразовании хрящей в кости, в частности, в длинные кости.
Ахондроплазия является одним из старейших известных врожденных дефектов. Частота заболевания ахондроплазией оценивается в диапазоне от 1 заболевшего на 10000 новорожденных в Латинской Америке до около 12 заболевших на 77000 новорожденных в Дании. Средний показатель по всему миру составляет примерно 1 заболевший на 25000 новорожденных.
К настоящему времени описано свыше 3500 наследственных болезней, около 5-5,5% детей рождаются с наследственной или врожденной патологией.
Наследственные болезни - заболевания, возникновение и развитие которых связано с изменениями (мутациями) генетического материала. В зависимости от характера мутаций выделяют моногенные наследственные, хромосомные, митохондриальные и мультифакториальные болезни. От наследственных заболеваний следует отличать врожденные заболевания, которые обусловлены внутриутробными повреждениями, вызванными, например, инфекцией (сифилис или токсоплазмоз) или воздействием иных повреждающих факторов на плод во время беременности.
Многие генетически обусловленные заболевания проявляются не сразу после рождения, а спустя некоторое, порой весьма долгое, время.
Наследственные болезни имеют свои особенности:
1. НБ часто носят семейный характер. В то же время наличие заболевания только у одного из членов родословной не исключает наследственного характера этой болезни (новая мутация, появление рецессивной гомозиготы).
2. При НБ в процесс вовлекаются сразу несколько органов и систем.
3. Для НБ характерно прогрессирующее хроническое течение.
4. При НБ бывают редко встречающиеся специфические симптомы или их сочетания: голубые склеры говорят о несовершенном остеогенезе, потемнение мочи на пеленках - об алкаптонурии, мышиный запах - о фенилкетонурии и др.
Этиология наследственных болезней . Этиологическими факторами наследственных болезней являются мутации (изменения) наследственного материала.
Мутации в хромосомах приводят к развитию хромосомных болезней, что может приводить к внутриутробной гибели эмбрионов и плодов, врожденным порокам развития и другим клиническим проявлениям. Чем больше хромосомного материала вовлечено в мутацию, тем раньше проявляется заболевание и тем значительнее нарушения в физическом и психическом развитии индивидуума. Известно около 1000 типов хромосомных нарушений, выявляемых у человека. Хромосомные заболевания редко передаются от родителей к детям, в основном это случайно возникшая новая мутация. Но около 5% людей являются носителями сбалансированных изменений в хромосомах, поэтому при бесплодии, мертворождениях, привычном невынашивании или наличии в семье ребенка с хромосомной патологией необходимо исследовать хромосомы каждого из супругов
Генными болезнями называются заболевания, обусловленные изменениями структуры молекулы ДНК (генные мутации).
Моногенные заболевания (собственно наследственные заболевания) - фенотипически генные мутации - могут проявляться на молекулярном, клеточном, тканевом, органном и организменном уровнях.
Полигенные заболевания (мультифакториальные) - болезни с наследственной предрасположенностью, обусловлены взаимодействием нескольких (или многих) генов и факторов окружающей среды.
Вклад наследственных и врожденных болезней в младенческую и детскую смертность в развитых странах (по материалам ВОЗ) велик . Среди главных причин смерти в возрасте до 1 года доля перинатальных факторов составляет 28%, врожденные и наследственные болезни -25%, синдром внезапной смерти ребенка - 22%, инфекции -9%, другие - 6%. Главными причинами смерти в возрасте от 1 года до 4 лет являются несчастные случаи (31%), врожденные и наследственные болезни (23%), опухоли (16%), инфекции (11%), другие (6%).
Доказана значительная роль наследственной предрасположенности в возникновении широко распространенных болезней (болезнь желудка и двенадцатиперстной кишки, эссенциальная гипертензия, ишемическая болезнь сердца, язвенная псориаз, бронхиальная астма и др.). Поэтому для профилактики и лечения этих болезней необходимо знать механизмы взаимодействия средовых и наследственных факторов в их возникновении и развитии.
Наследственные болезни длительное время не поддавались лечению, а единственным методом профилактики была рекомендация воздержаться от деторождения. Эти времена прошли. Современная медицинская генетика вооружила клиницистов методами ранней, досимптомной (доклинической) и даже пренатальной диагностики наследственных болезней. Интенсивно развиваются и в некоторых центрах уже применяются методы преимплантационной (до имплантации зародыша) диагностики.
Сейчас сложилась стройная система профилактики наследственных болезней: медико-генетическое консультирование, преконцепционная профилактика, пренатальная диагностика, массовая диагностика у новорожденных наследственных болезней обмена, поддающихся диетической и лекарственной коррекции, диспансеризация больных и членов их семей. Внедрение этой системы обеспечивает снижение частоты рождения детей с врожденными пороками развития и наследственными болезнями на 60-70%.
Моногенные болезни (МБ) или генные (так их называют за рубежом) заболевания. В основе МБ лежат единичные генные или точковые мутации. МБ составляют значительную долю наследственной патологии и насчитывают сегодня более 4500 заболеваний. По данным литературы, в разных странах они выявляются у 30-65 детей в расчете на 1000 новорожденных, что составляет 3,0-6,5%, а в структуре общей смертности детей до 5 лет на их долю приходится 10-14%. Заболевания многочислены и отличаются выраженным клиническим полиморфизмом. Генные болезни чаще всего проявляются наследственными дефектами обмена веществ - ферментопатиями. Одна и та же генная болезнь может быть обусловлена разными мутациями. Например, в гене муковисцидоза описано свыше 200 таких мутаций, в гене фенилкетонурии - 30. В некоторых случаях мутации в разных частях одного гена могут приводить к различным болезням (например, мутации RET-онкогена).
Патологические мутации могут реализовываться в разные периоды онтогенеза. Большая часть их проявляется внутриутробно (до 25% всей наследственной патологии) и в допубертатном возрасте (45%). Около 25% патологических мутаций проявляются в пубертатном и юношеском возрасте, и лишь 10% моногенных болезней развиваются в возрасте старше 20 лет.
Вещества, накапливающиеся в результате отсутствия или снижения активности ферментов, либо сами оказывают токсическое действие, либо включаются в цепи вторичных обменных процессов, в результате которых образуются токсические продукты. Общая частота генных болезней в популяциях людей составляет 2-4%.
Генные болезни классифицируются: согласно типам наследования (аутосомно-доминантные, аутосомно-рецессивные, Х-сцепленные доминантные и т.д.); по характеру метаболического дефекта - наследственные болезни обмена -НБО (болезни, связанные с нарушением аминокислотного, углеводного, липидного, минерального обменов, обмена нуклеиновых кислот и др.); в зависимости от системы или органа, наиболее вовлеченного в патологический процесс (нервные, глазные, кожные, эндокринные и др.).
Среди НБО выделяют:
Болезни аминокислотного обмена (ФКУ, тирозиноз, алкаптонурия, лейциноз и др.);
Болезни углеводного обмена (галактоземия, гликогенозы, мукополисахаридозы);
Болезни порфиринового и билирубинового обмена (синдромы Жильбера, Криглер-Найяра, порфирии и др.);
Болезни биосинтеза кортикостероидов (адрено-генитальный синдром, гипоальдостеронизм и др.);
Болезни пуринового и пирамидинового обмена (оротовая ацидурия, подагра и др.);
Болезни липидного обмена (эссенциальные семейные липидозы, ганглиозидозы, сфинголипидозы, цереброзидозы и др.);
Болезни эритрона (анемия Фанкони, гемолитические анемии, дефицит глюкозо-6-фосфатдегидрогеназы и др.);
Болезни обмена металлов (болезни Вильсона-Коновалова, Менкеса, семейный периодический паралич и др.);
болезни транспорта систем почек (болезнь де Тони-Дебре-Фанкони, тубулопатии, вита- мин D-резистентный рахит и др.).
Хромосомными болезнями (хромосомными синдромами) называются комплексы множественных врожденных пороков развития, вызываемых числовыми (геномные мутации) или структурными (хромосомные аберрации) изменениями хромосом, видимыми в световой микроскоп.
Хромосомные аберрации и изменения количества хромосом, как и генные мутации, могут возникать на разных этапах развития организма. Если они возникают в гаметах родителей, то аномалия будет наблюдаться во всех клетках развивающегося организма (полный мутант). Если аномалия возникает в процессе эмбрионального развития при дроблении зиготы, кариотип плода будет мозаичным. Мозаичные организмы могут содержать несколько (2, 3, 4 и более) клеточных клонов с различными кариотипами. Это явление может сопровождаться мозаицизмом во всех, либо в отдельных органах и системах. При незначительном количестве аномальных клеток фенотипические проявления могут не обнаруживаться.
Хромосомные болезни у новорожденных детей встречаются с частотой примерно 2,4 случая на 1000 родившихся. Большинство хромосомных аномалий (полиплоидии, гаплоидии, трисомии по крупным хромосомам, моносомий) несовместимы с жизнью - эмбрионы и плоды элиминируются из организма матери, в основном, в ранние сроки беременности.
Хромосомные аномалии возникают и в соматических клетках с частотой около 2%. В норме такие клетки элиминируются иммунной системой, если они проявляют себя чужеродно. Однако, в некоторых случаях (активация онкогенов) хромосомные аномалии могут быть причиной злокачественного роста. Например, транслокация между 9-й и 22-й хромосомами вызывает хронический миелолейкоз.
Общим для всех форм хромосомных болезней является множественность поражения. Это черепно-лицевые поражения, врожденные пороки развития систем органов, замедленные внутриутробные и постнатальные рост и развитие, отставание в психическом развитии, нарушения функций нервной, иммунной и эндокринной систем.
Фенотипические проявления хромосомных мутаций зависят от следующих главных факторов: особенностей вовлеченной в аномалию хромосомы (специфический набор генов), типа аномалии (трисомия, моносомия, полная, частичная), размера недостающего (при частичной моносомии) или избыточного (при частичной трисомии) генетического материала, степени мозаичности организма по аберрантным клеткам, генотипа организма, условий среды. В настоящее время выяснилось, что при хромосомных мутациях наиболее специфичные для того или иного синдрома проявления обусловлены изменениями небольших участков хромосом. Так, специфические симптомы болезни Дауна обнаруживаются при трисомии небольшого сегмента длинного плеча 21-й хромосомы (21q22.1), синдрома кошачьего крика - при делеции средней части короткого плеча 5-й хромосомы (5р15), синдрома Эдвардса - при трисомии сегмента длинного плеча хромосомы
Окончательный диагноз хромосомных болезней устанавливается цитогенетическими методами.
Трисомии. Наиболее часто у человека встречаются трисомии по 21-й, 13-й и 18-й паре хромосом.
Синдром (болезнь) Дауна (СД) - синдром трисомии 21 - самая частая форма хромосомной патологии у человека (1:750). Цитогенетически синдром Дауна представлен простой трисомией (94% случаев), транслокационной формой (4%) или мозаицизмом (2% случаев). У мальчиков и девочек патология встречается одинаково часто.
Достоверно установлено, что дети с синдромом Дауна чаще рождаются у пожилых родителей. Возможность возникновения повторного случая заболевания в семье с трисомией хромосомы 21 составляет 1-2% (с возрастом матери риск увеличивается). Три четверти всех случаев транслокаций при болезни Дауна обусловлены мутацией de novo. 25% случаев транслокации носят семейный характер, при этом возвратный риск гораздо выше (до 15%) и во многом зависит от того, кто из родителей несет симметричную транслокацию и какая из хромосом вовлечена.
Для больных характерны: округлой формы голова с уплощенным затылком, узкий лоб, широкое, плоское лицо, типичны эпикант, гипертелоризм, запавшая спинка носа, косой (монголоидный) разрез глазных щелей, пятна Брушфильда (светлые пятна на радужке), толстые губы, утолщенный язык с глубокими бороздами, выступающий изо рта, маленькие, округлой формы, низко расположенные ушные раковины со свисающим завитком, недоразвитая верхняя челюсть, высокое небо, неправильный рост зубов, короткая шея.
Из пороков внутренних органов наиболее типичны пороки сердца (дефекты межжелудочковой или межпредсердной перегородок, фиброэластоз и др.) и органов пищеварения (атрезия двенадцатиперстной кишки, болезнь Гиршпрунга и др.). Среди больных с синдромом Дауна с более высокой частотой, чем в популяции, встречаются случаи лейкемии и гипотиреоза. У маленьких детей резко выражена мышечная гипотония, а у детей старшего возраста часто обнаруживается катаракта. С самого раннего возраста отмечается отставание в умственном развитии. Среднее значение IQ составляет 50, но чаще встречается умеренная задержка умственного развития. Средняя продолжительность жизни при синдроме Дауна значительно ниже (36 лет), чем в популяции.
Синдром Патау (СП ) - синдром трисомии 13 - встречается с частотой 1:7000 (с учетом мертворождений). Имеются два цитогенетических варианта синдрома Патау: простая трисомия и робертсоновская транслокация. 75% случаев трисомии хромосомы 13 обусловлено появлением дополнительной хромосомы 13. Между частотой возникновения синдрома Патау и возрастом матери прослеживается зависимость, хотя и менее строгая, чем в случае болезни Дауна. 25% случаев СП - следствие транслокации с вовлечением хромосом 13-й пары, в том числе в трех из четырех таких случаев мутация de novo. В четверти случаев транслокация с вовлечением хромосом 13-й пары имеет наследственный характер с возвратным риском 14%.
При СП наблюдаются тяжелые врожденные пороки. Дети с синдромом Патау рождаются с массой тела ниже нормы (2500 г). У них выявляются: умеренная микроцефалия, нарушение развития различных отделов ЦНС, низкий скошенный лоб, суженные глазные щели, расстояние между которыми уменьшено, микрофтальмия и колобома, помутнение роговицы, - запавшая переносица, широкое основание носа, деформированные ушные раковины, расщелина верхней губы и неба, полидактилия, флексорное положение кистей, короткая шея.
У 80% новорожденных встречаются пороки развития сердца: дефекты межжелудочковой и межпредсердной перегородок, транспозиции сосудов и др. Наблюдаются фиброкистозные изменения поджелудочной железы, добавочные селезенки, эмбриональная пупочная грыжа. Почки увеличены, имеют повышенную дольчатость и кисты в корковом слое, выявляются пороки развития половых органов. Для СП характерна задержка умственного развития.
Большинство больных с синдромом Патау (98%) умирают в возрасте до года, оставшиеся в живых страдают глубокой идиотией.
Синдром Эдвардса (СЭ) - синдром трисомии 18 - встречается с частотой примерно 1:7000 (с учетом мертворождений). Дети с трисомией 18 чаще рождаются у пожилых матерей, взаимосвязь с возрастом матери менее выражена, чем в случаях трисомии хромосомы 21 и 13. Для женщин старше 45 лет риск родить больного ребенка составляет 0,7%. Цитогенетически синдром Эдвардса представлен простой трисомией 18 (90%), в 10% случаев наблюдается мозаицизм. У девочек встречается значительно чаще, чем у мальчиков, что связано, возможно, с большей жизнестойкостью женского организма.
Дети с трисомией 18 рождаются с низким весом (в среднем 2177 г), хотя сроки беременности нормальные или даже превышают норму.
Фенотипические проявления синдрома Эдвардса многообразны: часто отмечаются аномалии мозгового и лицевого черепа, мозговой череп долихоцефалической формы, нижняя челюсть и ротовое отверстие маленькие, глазные щели узкие и короткие, ушные раковины деформированы и в подавляющем большинстве случаев расположены низко, несколько вытянуты в горизонтальной плоскости, мочка, а часто и козелок отсутствуют; наружный слуховой проход сужен, иногда отсутствует, грудина короткая, из-за чего межреберные промежутки уменьшены и грудная клетка шире и короче нормальной, аномальное развитие стопы: пятка резко выступает, свод провисает (стопа-качалка), большой палец утолщен и укорочен; отмечаются пороки сердца и крупных сосудов: дефект межжелудочковой перегородки, аплазии одной створки клапанов аорты и легочной артерии, гипоплазия мозжечка и мозолистого тела, изменения структур олив, выраженная умственная отсталость, снижение мышечного тонуса, переходящее в повышение со спастикой.
Продолжительность жизни детей с синдромом Эдвардса невелика: 60% детей умирают в возрасте до 3 мес., до года доживает лишь один ребенок из десяти; оставшиеся в живых - глубокие олигофрены.
Синдром трисомии X. Частота встречаемости 1:1000. Кариотип 47, ХХХ. В настоящее время имеются описания тетра-и пентосомий X. Трисомия по Х-хромосоме возникает в результате нерасхождения половых хромосом в мейозе или при первом делении зиготы.
Синдрому полисомии X присущ значительный полиморфизм. Женский организм с мужеподобным телосложением. Могут быть недоразвиты первичные и вторичные половые признаки. В 75% случаев у больных наблюдается умеренная степень умственной отсталости. У некоторых из них нарушена функция яичников (вторичная аменорея, дисменорея, ранняя менопауза). Иногда такие женщины могут иметь детей. Повышен риск заболевания шизофренией. С увеличением числа дополнительных Х-хромосом нарастает степень отклонения от нормы.
Синдром Шерешевского-Тернера (моносомия X). Частота встречаемости 1:1000.
Кариотип 45,Х. У 55% девочек с этим синдромом обнаруживается кариотип 45,X, у 25% - изменение структуры одной из Х-хромосом. В 15% случаев выявляется мозаичность в виде двух или более клеточных линий, одна из которых имеет кариотип 45,X, а другая представлена кариотипами 46,XX или 46,XY. Третья клеточная линия наиболее часто представлена кариотипом 45,Х, 46^ХХ, 47,ХХХ. Риск наследования синдрома составляет 1 случай на 5000 новорожденных. Фенотип женский.
У новорожденных и детей грудного возраста отмечаются признаки дисплазии (короткая шея с избытком кожи и крыловидными складками, лимфатический отек стоп, голеней, кистей рук и предплечий, вальгусная деформация стоп, множественные пигментные пятна, низкорослость. В подростковом возрасте выявляются отставание в росте (рост взрослых 135-145 см) и в развитии вторичных половых признаков. Для взрослых характерно: низкое расположение ушных раковин, недоразвитие первичных и вторичных половых признаков, дисгенезия гонад, сопровождающаяся первичной аменореей, у 20% больных имеются пороки сердца (коарктация аорты, стеноз аорты, пороки развития митрального клапана),у 40% - пороки почек (удвоение мочевыводящих путей, подковообразная почка).
У больных, имеющих клеточную линию с Y-хромосомой, может развиться гонадобластома, часто наблюдается аутоиммунный тиреоидит. Интеллект страдает редко. Недоразвитие яичников приводит к бесплодию. Для подтверждения диагноза наряду с исследованием клеток периферической крови проводятся биопсия кожи и исследование фибробластов. В некоторых случаях генетическое исследование позволяет выявить синдром Нунан, который имеет схожие фенотипические проявления, однако этиологически не связан с синдромом Шерешевского-Тернера. В отличие от последнего при синдроме Нунан заболеванию подвержены как мальчики, так и девочки, а в клинической картине доминирует задержка умственного развития, характерен Тернер-фенотип при нормальном мужском или женском кариотипе. У большинства больных синдромом Нунан имеется нормальное половое развитие и сохранена фертильность. В большинстве случаев заболевание не сказывается на продолжительности жизни пациентов.
Синдром Клайнфелтера. Частота встречаемости 1: 1000 мальчиков. Кариотип 47,XXY. У 80% мальчиков с синдромом Клайнфелтера, в 20% случаев обнаруживается мозаицизм, при котором одна из клеточных линий имеет кариотип 47,XXY. Возвратный риск для синдрома Клайнфелтера не превышает общепопуляционные показатели и составляет 1 случай на 2000 живорожденных детей. Фенотип мужской.
Клиника отличается широким разнообразием и неспецифичностью проявлений. У мальчиков с этим синдромом рост превышает средние показатели, характерные для данной семьи, у них длинные конечности, женский тип телосложения, гинекомастия. Слабо развит волосяной покров, снижен интеллект. Вследствие недоразвития семенников слабо выражены первичные и вторичные половые признаки, нарушено течение сперматогенеза. Половые рефлексы сохранены. Иногда эффективно раннее лечение мужскими половыми гормонами. Чем больше в наборе Х-хромосом, тем значительнее снижен интеллект. Инфантильность и поведенческие проблемы при синдроме Клайнфелтера создают трудности социальной адаптации.
Иногда возможны случаи увеличения количества Y-хромосом: XYY, XXYY и др. При этом больные имеют признаки синдрома Клайнфелтера, высокий рост (в среднем 186 см) и агрессивное поведение. Могут быть аномалии зубов и костной системы. Половые железы развиты нормально. Чем больше в наборе Y-хромосом, тем значительнее снижение интеллекта агрессивность поведения.
Помимо полных трисомий и моносомий известны синдромы, связанные с частичными трисомиями и моносомиями практически по любой хромосоме. Однако, эти синдромы встречаются реже одного случая на 100 000 рождений.
Диагностика НБ . В клинической генетике для диагностики различных форм наследственной патологии применяются: клинико-генеалогический метод, специальные и дополнительные (лабораторные, инструментальные) методы исследования.
Медико-генетическое консультирование . Основная цель медико-генетические консультирования - информировать заинтересованных лиц о вероятности риска появления в потомстве больных. К медико-генетическим мероприятиям относится также пропаганда генетических знаний среди населения, т.к. это способствует более ответственному подходу к деторождению. Медико-генетическая консультация воздерживается от мер принудительного или поощрительного характера в вопросах деторождения или вступления в брак, принимая на себя лишь функцию информации.
Медико-генетическое консультирование (МГК) - это специализированная помощь населению по предупреждению появления в семье больных с наследственной патологией, по выявлению, консультированию больных с НБ, информированию населения о НБ, а также способах его предупреждения и лечения.
Основные задачи МГК:
Установление точного диагноза наследственного заболевания и определение типа наследования заболевания в данной семье;
Составление прогноза рождения ребенка с наследственной болезнью, расчет риска повторения болезни в семье;
Определение наиболее эффективного способа профилактики, помощь семье в принятии правильного решения;
Пропаганда медико-генетических знаний среди врачей, населения.
Показания для МГК:
Задержка физического развития; карликовый рост (не более 140 см для взрослых), врожденные деформация верхних и/или нижних конечностей, пальцев, позвоночника, грудной клетки, черепа, деформация лица, изменение числа пальцев на руках и ногах, синдактилия, комбинации врожденных уродств, врожденная ломкость костей;
Задержка полового развития, неопределенный пол; недоразвитие НПО и вторичных половых признаков;
Задержка умственного развития, умственная отсталость, врожденная глухота или глухонемота;
Повышенное количество стигм дизэмбриогенеза;
Множественные пороки развития или сочетание изолированных пороков и малых аномалий развития;
Атрофия мышц, гипертрофия мышц, спастическое подергивание мышц, насиль-ственные движения, параличи, нетравматическая хромота, нарушение походки, неподвижность или тугоподвижность в суставах;
Слепота, микрофтальм, врожденная катаракта, врожденная глаукома, колобомы, аниридия, нистагм, птоз, прогрессирующее ухудшение сумеречного зрения;
Сухость или усиленное ороговение кожи ладоней и подошв, других участков тела, пятна коричневого цвета и множественные опухоли на коже, спонтанное или индуцированное образование пузырей, отсутствие ногтей, алопеция, непрорезывание зубов;
Хронические прогрессирующие заболевания неясного генеза;
Резкое ухудшение состояния после кратковременного периода нормального развития ребенка. Бессимптомный промежуток может составлять от нескольких часов до недель и зависит от природы дефекта, режима питания и других факторов;
Вялость или, наоборот повышенный тонус и судороги у новорожденного, непрекращающаяся рвота у новорожденного, прогрессирующие неврологические расстройства;
Необычный запах тела и/или мочи («сладкий», «мышиный», «вареной капусты», «потных ног») и др.;
Наличие в семье наследственной патологии, пороков развития, сходные случаи заболевания в семье, случаи внезапной смерти ребенка в раннем возрасте;
Бесплодие, привычное невынашивание, мертворождения;
Кровнородственный брак
Еще до планирования деторождения, а также при рождении больного ребенка (ретроспективно), каждая супружеская пара должна пройти медико-генетическое консультирование.
Этапы МГК:
1. Верификация клинического диагноза наследственного (или предположительно
наследственного).
2. Установление характера наследования заболевания в консультируемой семье.
3. Оценка генетического риска повторения заболевания (генетический прогноз).
4. Определение способов профилактики.
5. Объяснение обратившимся смысла собранной и проанализированной медико-генетическойинформации.
Методы пренаталъной диагностики наследственных болезней . Пренатальная диагностика связана с решением ряда биологических и этических проблем до рождения ребенка, так как при этом речь идет не об излечении болезни, а о предупреждении рождения ребенка с патологией, не поддающейся лечению (обычно путем прерывания беременности с согласия женщины и проведения перинатального консилиума). При современном уровне развития пренатальной диагностики можно установить диагноз всех хромосомных болезней, большинства врожденных пороков развития, энзимопатий, при которых известен биохимический дефект. Часть из них можно установить практически на любом сроке беременности (хромосомные болезни), часть - после 11-12-й недели (редукционные пороки конечностей, атрезии, анэнцефалию), часть - только во второй половине беременности (пороки сердца, почек, ЦНС).
Показания для пренатальной диагностики:
Наличие в семье точно установленного наследственного заболевания;
Возраст матери старше 37 лет;
Носительство матерью гена Х-сцепленного рецессивного заболевания;
Наличие в анамнезе у беременных спонтанных абортов в ранние сроки беременности, мертворождений неясного генеза, детей с множественными пороками развития и с хромосомной патологией;
Наличие структурных перестроек хромосом (особенно транслокаций и инверсий) у одного из родителей;
Гетерозиготность обоих родителей по одной паре аллелей при патологии с аутосомно-рецессивным типом наследования;
Беременные из зоны повышенного радиационного фона.
Дальнейшее развитие и распространение методов пренатальной диагностики наследственных заболеваний позволят значительно снизить частоту наследственной патологии новорожденных.
Неонатальный скрининг. В рамках реализуемого Приоритетного национального проекта «Здоровье» предусмотрено расширение неонатального скрининга и сейчас проводится скриниг на фенилкетонурию, врожденный гипотиреоз, адреногенитальный синдром, галактоземию, муковисцидоз. Массовое обследование новорожденных (неонатальный скрининг) на НБО является основой профилактики наследственных болезней в популяциях. Неонатальная диагностика наследственных заболеваний позволяет определить распространенность заболевания на конкретной территории, в конкретном субъекте Российской Федерации и в целом по стране, обеспечить раннее выявление детей, страдающих наследственными заболеваниями и своевременно начать лечение, предупредить инвалидность и развитие тяжелых клинических последствий, снизить детскую смертность от наследственных заболеваний, выявить семьи, нуждающиеся в генетической консультации, с целью предупреждения рождения детей с данными наследственными заболеваниями.
Результаты скрининга новорожденных на наследственные болезни обмена в Чувашской Республике за период с 1999-2008 гг.
Лечение наследственных заболеваний . Несмотря на большие успехи в совершенствовании цитогенетических, биохимических и молекулярных методов изучении этиологии и патогенеза НЗ, по-прежнему, основным остается симптоматическое лечение, которое мало отличается от лечения любых других хронических заболеваний. И все же, в настоящее время в арсенале врачей-генетиков находится немало средств патогенетического лечения; в первую очередь это касается наследственных болезней обмена (НБО). Клинические проявления при НБО являются итогом нарушений в цепи превращений (метаболизма) продуктов (субстратов) в организме человека; мутация гена приводит к дефектной работе ферментов и коферментов. Патогенетическая терапия разработана примерно для 30 НБО. Существует несколько направлений терапии НБО:
1. Диетотерапия. Ограничение или полное прекращение поступления в организм продуктов, метаболизм которых нарушен в результате ферментативного блока. Этот прием используется в случаях, когда избыточное накопление субстрата оказывает токсичное действие на организм. Иногда (особенно когда субстрат не является жизненно необходимым и может синтезироваться в достаточном количестве обходными путями) такая диетотерапия оказывает очень хороший эффект. Типичный пример - галактоземия. Несколько сложнее дело обстоит при фенилкетонурии. Фенилаланин - незаменимая аминокислота, поэтому ее нельзя полностью исключать из пищи, а надо индивидуально подбирать для больного физиологически необходимую дозу фенилаланина. Также диетотерапия разработана для тирозинемии, лейциноза наследственной непереносимости фруктозы, гомоцистинурии и т.д.
2. Восполнение коферментов. При ряде НБО изменяется не количество необходимого фермента, а его структура, в результате чего нарушается связывание с коферментом, наступает метаболический блок. Чаще всего речь идет о витаминах. Дополнительное введение больному коферементов (чаще определенных доз витаминов) дает положительный эффект. В качестве таких «помощников» используют пиридоксин, кобаламин, тиамин, препараты карнитина, фолаты, биотин, рибофлавин и т.д.
3. Усиленное выведение токсических продуктов, накапливающихся в случае блокирования их дальнейшего метаболизма. К числу таких продуктов относится, например, медь при болезни Вильсона-Коновалова (для нейтрализации меди больному вводят D-пеницилламин), железо при гемоглобинопатиях (для предотвращения гемосидероза паренхиматозных органов назначают десферал.
4. Искусственное введение в организм больного продукта блокированной у него реакции. Например, прием цитидиловой кислоты при оротоацидурии (заболевание, при котором страдает синтез пиримидинов) устраняет явления мегалобластической анемии.
5. Воздействие на «испорченные» молекулы. Этот метод применяется для лечения серповидно-клеточной анемии и направлен на уменьшение вероятности образования кристаллов гемоглобина 3. Ацетилсалициловая кислота усиливает ацетилирование HbS и таким путем снижает его гидрофобность, обусловливающую агрегацию этого белка.
6. Возмещение отсутствующего фермента. Такой метод успешно применяется при лечении адрено-генитального синдрома (введение стероидных гормонов с глюко- и минералокортикоидной активностью), гипофизарного нанизма (введение гормона роста), гемофилии (антигемофильный глобулин). Однако, для эффективного лечения необходимо знать все тонкости патогенеза заболевания, его биохимических механизмов. Новые успехи на этом пути свзаны с достижениями физико-химической биологии, генной инженерии и биотехнологии.
7. Блокирование патологической активности ферментов с помощью специфических ингибиторов или конкурентное торможение аналогами субстратов данного фермента. Этот метод лечения применяется при избыточной активации систем свертывания крови, фибринолиза, а также при освобождении из разрушенных клеток лизосомальных ферментов.
Все большее применение в лечении НЗ находит трансплантация клеток, органов и тканей. Тем самым в организм больного вместе с органом или тканью вводится нормальная генетическая информация, обеспечивающая правильные синтез и работу ферментов и предохраняющая организм от последствий произошедшей мутации. Аллотрансплантация используется для лечения: синдромов Ди Джорджи (гипоплазия тимуса и паращитовидных желез) и Незелофа - пересадка тимуса; остеопетроза рецессивного, мукополисахаридозов, болезни Гоше, анемии Фанкони - пересадка костного мозга; первичных кардиомиопатий - пересадка сердца; болезни Фабри, амилоидозе, синдроме Альпорта, наследственного поликистоза почек - пересадка почек и т.д.
Врожденная патология в виде врожденных пороков развития может возникнуть в критические периоды внутриутробного развития под действием факторов внешней среды (физических, химических, биологических и др.). При этом поражения или изменения генома нет.
Факторами риска рождения детей с пороками развития различного генеза могут быть: возраст беременной старше 36 лет, предшествующие рождения детей с пороками развития, самопроизвольные аборты, кровнородственный брак, соматические и гинекологические заболевания матери, осложненное течение беременности (угроза прерывания беременности, недоношенность, переношенность, тазовое предлежание, мало- и многоводие).
Отклонения в развитии органа или системы органов могут быть грубыми с выраженной функциональной недостаточностью или всего лишь косметическим дефектом. Врожденные пороки развития обнаруживаются в периоде новорожденности. Небольшие отклонения в строении, которые в большинстве случаев не влияют на нормальную функцию органа, называют аномалиями развития или стигмами дисэмбриогенеза.
Стигмы обращают на себя внимание в тех случаях, когда их насчитывается более 7 у одного ребенка, в этом случае можно констатировать диспластическую конституцию. Имеются трудности в клинической оценке диспластической конституции, так как одна или несколько стигм могут оказаться:
1. вариантом нормы;
2. симптомом заболевания;
3. самостоятельным синдромом.





